 2026-04-07
2026-04-07 477
477 来源: advanced science
来源: advanced science

Adv Sci|复旦、上交大学研究团队发现组蛋白去乙酸化酶(HDACs)诱导的STAT3脱乙酰化对DLBCL中M2巨噬细胞积累至关重要
肿瘤相关巨噬细胞(TAM)是免疫抑制微环境的重要组成部分,已被指出是弥漫性大B细胞淋巴瘤(DLBCL)复发的关键因素。然而,调控DLBCL-TAMs的分子机制及潜在干预机制尚未明确。
2026年3月31日,复旦大学陈彤及上海交通大学童茵共同通讯在Advance Science上发表题为"Spatiotemporal Sequential Delivery of Chidamide Regulates Macrophage Reprogramming in Lymphoma Microenvironment Through HDACs-STAT3 Pathway"的论文,该研究发现组蛋白去乙酸化酶(HDACs)诱导的STAT3脱乙酰化对DLBCL中M2巨噬细胞积累至关重要。

鉴于HDAC抑制剂chidamide在DLBCL治疗中的不可忽视的不良反应,研究团队开发了一种结合肽修饰胞外囊泡(M2pep-EVs)的M2靶向递送系统,以实现最佳的瘤内递送。结合pH响应的水凝胶TSPBA/PVA,将chidamide装载于M2pep-EVs中,并在酸性淋巴瘤微环境中智能释放为原位Chid@M2pep-EV。通过向M2巨噬细胞的靶向递送,chidamide充分抑制了HDACs,增强了STAT3的乙酰化,将M2比例重新编程为M1表型,最终抑制了体内淋巴瘤的生长。通过降低剂量和不良反应,Chid@M2pep-EVs系统为治疗难治性/复发性淋巴瘤提供了新的转化策略。
非霍奇金淋巴瘤(NHL)是一类异质性淋巴恶性肿瘤,主要起源于B细胞、T细胞和自然杀伤细胞。其中弥漫性大B细胞淋巴瘤(DLBCL)属于侵袭性亚型,其中30%–40%的患者会复发。新兴证据表明,肿瘤相关巨噬细胞(TAM),尤其是M2表型,在疾病进展和治疗耐药过程中创造了免疫抑制微环境。临床研究显示,M2巨噬细胞的肿瘤富集与患者生存率下降相关及CAR-T细胞疗效降低,而实验模型则直接证明M2表型在介导化疗耐药中起作用。然而,DLBCL微环境中M2极化的分子驱动因素尚未明确,阻碍了进一步靶向干预的发展。
Chidamide(Chid)是一种选择性组蛋白去乙酰化酶(HDAC)抑制剂,获批用于T细胞淋巴瘤治疗,具有双重抗肿瘤机制:高剂量Chid直接诱导恶性细胞凋亡,而低剂量Chid调节免疫反应,包括抑制M2极化。尽管高剂量方案在临床使用中占主导地位,因其更优越的反应率,但剂量相关的严重毒性(如骨髓抑制、器官功能障碍)也不可忽视。相反,低剂量免疫调节受限于药物动力学不良和副作用。因此,开发策略以增强Chid肿瘤特异性递送,特别是对重编程的M2巨噬细胞,有望在治疗效果与安全性之间取得平衡。
细胞外囊泡(EVs),即内源性纳米尺度载体(30–150 nm),为靶向药物递送提供了理想的解决方案。其生物相容性、生物分布多样性以及包裹疏水或亲水货物的能力已被应用于从纤维化到心血管疾病等多种疾病。然而,EV在淋巴瘤治疗中的应用尚未被充分探索。一个关键障碍是EV释放缺乏时空控制,可能导致爆发释放和剂量不灵活。为此,我们将EV与pH响应型水凝胶TSPBA/PVA(TP)结合,利用酸性淋巴瘤微环境实现持续的Chid释放,同时减轻全身毒性。
先前研究报告称,HDAC能够修饰STAT3以调控巨噬细胞重编程,而Chid在调节巨噬细胞免疫功能中的作用尚不明确。我们鉴定了STAT3脱乙酰化作为DLBCL中M2极化的关键开关,并开发了针对特定Chid给药的pH响应药物递送平台。先进结合M2靶向Chid与EV介导的递送,增强了现场抗肿瘤疗效并减少副作用,并在疾病进展期间通过TP水凝胶控制释放动力学(方案1)。我们的策略提供了一个范式,既能破坏免疫抑制生态位,又能增强淋巴瘤治疗的内在药物活性。
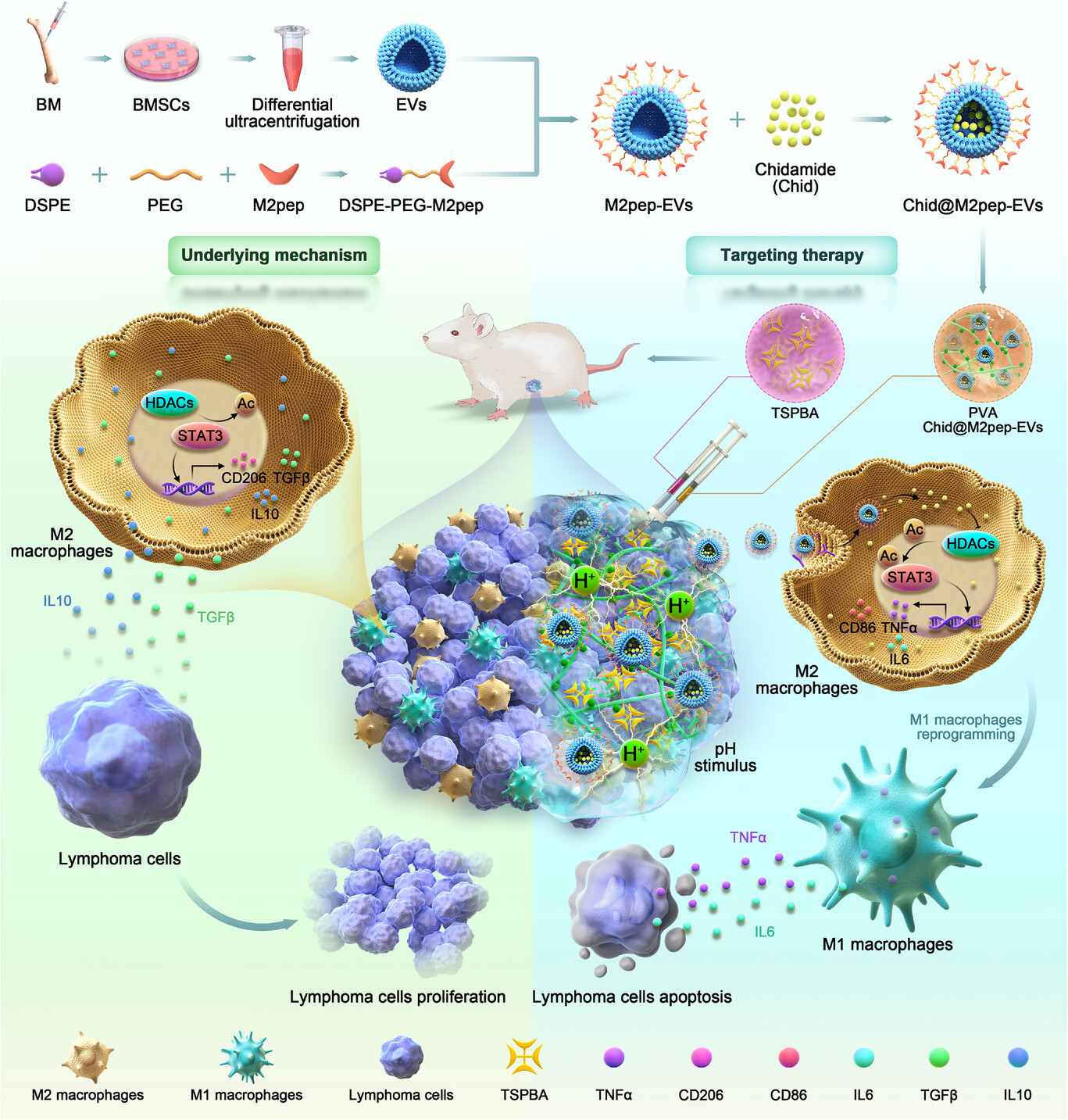
以往研究报道了淋巴瘤中M2巨噬细胞的招募。然而,目前尚无研究专门针对这些基因进行特定干预。在我们的研究中,通过结合电子凝胶、水凝胶和Chid,首次证明了靶向M2巨噬细胞促进其肿瘤细胞毒性重编程的可行性。Chid@M2pep-胚血/特种质检测显著抑制淋巴瘤生长,表现为M1巨噬细胞增加,M2巨噬细胞减少,淋巴瘤凋亡增强,较对照组更显著。未观察到重大器官毒性,解决了高剂量Chid的安全性问题,并验证了其在复发/难治性DLBCL中的转化潜力。
在机制上,Chid治疗下STAT3乙酰化水平升高,从而促进M1巨噬细胞相关基因的表达。M1巨噬细胞比例的增加促进了抗淋巴瘤疗效。先前研究报告称,STAT3去乙酰化促进M2巨噬细胞极化。我们的发现补充了这一发现,突出了STAT3乙酰化在调控巨噬细胞重编程中的独特作用。基于这些见解,我们开发了一种潜在的淋巴瘤治疗策略。
总之,本研究确定HDACs-STAT3-M2轴为DLBCL免疫抑制的驱动因子,并开发了一种靶向、pH响应的递送系统Chid@M2pep-EVs/TP,通过HDAC1/2/3抑制和STAT3乙酰化,高效将M2巨噬细胞重编程为M1表型,以抑制DLBCL生长。Chid@M2pep-EVs/TP为复发/难治性DLBCL治疗提供了一种新范式,具有对其他M2富集肿瘤恶性肿瘤的适应性潜力。
原文参考:https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202502791
备注:图片来源网络如有侵权请及时联系删除!

